New active substances and global marketing authorisations
Data and marketing exclusivity (otherwise known as regulatory data protection) prevents the use of pre-clinical and clinical data produced for the authorisation of an earlier reference product, for the subsequent purpose of approval of a generic version of that reference product. The bar against reference to the data lasts for eight years, but marketing of the generic is prevented for ten years1. As we explain in "There is more than one way to extend data exclusivity periods" there are a number of ways in which this exclusivity period can be extended, including for: new therapeutic indications, change of classification, orphan drugs.
But what exclusivity protection is available to the holder of the marketing authorisation for line extensions that may follow its original reference authorisation? Is every modification to a product entitled to a new data exclusivity period? This question is addressed by the concept of the "global marketing authorisation" provided in Directive 2001/83/EC, Article 6(1), second subparagraph:
When a medicinal product has been granted an initial marketing authorisation in accordance with the first sub-paragraph, any additional strengths, pharmaceutical forms, administration routes, presentations, as well as any variations and extensions shall also be granted an authorisation in accordance with the first sub-paragraph or be included in the initial marketing authorisation. All these marketing authorisations shall be considered as belonging to the same global marketing authorisation, in particular for the purpose of the application of Article 10(1).
As the European Commission's Notice to Applicants2 goes on to explain:
Thus, the global marketing authorisation contains the initial authorisation and all variations and extensions thereof, as well as any additional strengths, pharmaceutical form, administration routes or presentations authorised through separate procedures and under a different name, granted to the marketing authorisation holder of the initial authorisation. Where a product is initially authorised nationally and, subsequently, an additional strength, pharmaceutical form, administration route or presentation is authorised through the centralised procedure, this shall also be part of the same global marketing authorisation.
 However, the list of products described as falling within the global marketing authorisation lacks clarity, particularly as regards the meaning of pharmaceutical forms. The Notice to Applicants supplements this list using the concept of the "new active substance"3. A product will be assessed as a new active substance if it is:
However, the list of products described as falling within the global marketing authorisation lacks clarity, particularly as regards the meaning of pharmaceutical forms. The Notice to Applicants supplements this list using the concept of the "new active substance"3. A product will be assessed as a new active substance if it is:
- A chemical, biological or radiopharmaceutical substance not previously authorised as a medicinal product in the EU.
- An isomer, mixture of isomers, a complex or derivative or salt of a chemical substance previously authorised as a medicinal product in the European Union but differing significantly in properties with regard to safety and efficacy from that chemical substance previously authorised.
- A biological substance previously authorised as a medicinal product in the European Union, but differing in molecular structure, nature of the source material or manufacturing process.
- A radiopharmaceutical substance which is a radionucleotide, or a ligand not previously authorised as a medicinal product in the European Union, or the coupling mechanism to link the molecule and the radionucleotide has not been authorised previously in the European Union.
Unless such a substance can be shown to be "differing significantly in properties with regard to safety and efficacy" to the reference product there is a presumption that it is a generic medicinal product that is not entitled to an additional period of data exclusivity protection. But, how is this requirement to be satisfied? Furthermore, what exactly do "complex" and "derivative" mean?
The EMA has issued guidance in a "Reflection paper" on the considerations given specifically to whether an enantiomer, a complex, a derivative, or a different salt or ester should be designated as a new active substance in relation to the relevant reference active substance4. This covers:
- Criteria to be applied in deciding on whether an active substance differs significantly with regard to efficacy and/or safety compared to the relevant reference active substance
- Type of evidence required to show differences; and
- What might constitute a significant difference in safety and/or efficacy to justify new active substance status (evidence likely to be sufficient and evidence unlikely to be sufficient).
There are examples that illustrate how the Committee for Medicinal Products for Human Use of the European Medicines Agency (CHMP) will decide in certain circumstances. A leading example is eszopliclone. Here, despite a re-examination, the CHMP decided that Sepracor's active substance, the enantiomer eszopliclone, could not be regarded as a new active substance within the meaning of the legislation5. This was based on a comparison with its corresponding racemate zopliclone, which had already been on the market for many years.
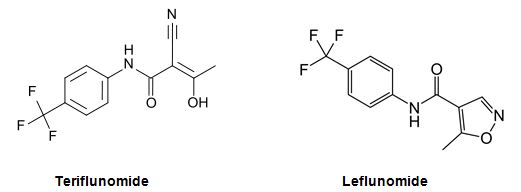
However, in another case, concerning aubagio (teriflunomide), the CHMP changed its opinion after re-examination to conclude that teriflunomide is a new active substance as compared to leflunomide. Teriflunomide is an active metabolite of leflunomide. In support of its decision the CHMP cites "differences between teriflunomide and leflunomide as regards safety".6
Another case in which the status of an active ingredient was re-examined by the CHMP is that of tecfidera (methyl fumarate). This was considered to be different from fumiderm, a combination of dimethyl fumarate, the calcium salt of ethyl fumarate, the magnesium salt of ethyl hydrogen fumarate and the zinc salt of ethyl hydrogen fumarate. However it is not clear how much this decision has to do with the fact that fumiderm had not previously been the subject of a European Union wide marketing authorisation rather than the compound being only one element on earlier combination7.
If you have any questions on this article or would like to propose a subject to be addressed by Synapse please contact us.
1 In the case of reference products for which authorisation was applied for after November 2005.
2 Volume 2A, Chapter 1, Section 2.3 (European Commission, November 2005).
3 Volume 2A, Chapter 1, Annex III (European Commission, November 2005).
4 EMA/651649/2010, dated 18 October 2012.
5 See General Court Case T-275/09 and Court of Justice Case C-477/11.

Related page


Paul England
Paul is a senior associate and professional support lawyer in the Patents group based in our London office.