Towards a harmonised EU assessment of the added therapeutic value of medicines?
September 2015
Policy department A of the European Parliament has produced for the Committee on Environment, Public Health and Food Safety (ENVI) a study which investigates the possibility of a harmonised EU approach concerning the assessment of the added therapeutic value (ATV) of medicinal products. The study reviews the current EU legal and policy framework and looks at the state-of-play within all 28 Member States. The study also contains policy recommendations on how a possible European harmonisation of the ATV assessment might be taken forward.
Background
About two-thirds of the EU's pharmaceutical expenditure is covered by public payers, so the pricing and reimbursement measures adopted by national health authorities have a significant impact on whether most patients will have access to a new medicinal product. National authorities in charge of pricing and reimbursement assess ATV-aspects; in particular they consider efficacy, safety and pharmaceutical quality of a medicinal product as well as its effectiveness, cost-effectiveness, budgetary impact, the severity of the disease and other factors.
Since ATV-based evaluations are the basis for national pharmaceutical pricing, the decision on whether or not a drug offers an additional benefit determines its price. In the current legal situation, pharmaceutical enterprises are required to carry out such assessments in each Member State in order to determine additional pharmaceutical benefits. This leads to two main problems. Firstly, pharmaceutical companies as well as national authorities have to invest increased efforts and expenditure, since the current systems do not provide for references to decisions in other EU Member States. Secondly, the decisions on a drug's benefits may vary from country to country, which impacts upon subsequent national pricing. Through the external price referencing principle that is applied by most European countries, national regulatory pricing influences pricing in other Member States. External price referencing means that the drug prices in the relevant countries are compared in order to fix a national price or establish a basis for price negotiations with the pharmaceutical undertaking. However, the legal requirements for ATV-based evaluations are not harmonised throughout the European Union.
 The study now investigates the possibility of a harmonised EU approach concerning the assessment of ATV of medicinal products. The policy departments are responsible for providing expertise and policy advice to support the activities of various European parliamentary bodies, most notably the committees. Based on analyses carried out either in-house or externally, policy departments provide information in all areas of Parliament's activities. The study was undertaken upon request of the ENVI Committee of the European Parliament. The aim was to investigate the feasibility and opportunity of introducing a harmonised EU approach concerning the assessment of the added therapeutic value of medicines in the European Union. The study sets out the general background to ATV and its use by EU Member States, and explains the different stages in the national decision-making process where ATV considerations play a role. Next, it provides a (non-exhaustive) mapping of practices and the current state-of-play in the 28 EU Member States, as well as an in-depth analysis of the use of ATV in six selected countries: Austria, France, Italy, Poland, Slovakia and Sweden. Based on the information and data collected, the study also provides legal and policy recommendations for streamlining the various national systems in place for ATV assessment towards a common EU approach.
The study now investigates the possibility of a harmonised EU approach concerning the assessment of ATV of medicinal products. The policy departments are responsible for providing expertise and policy advice to support the activities of various European parliamentary bodies, most notably the committees. Based on analyses carried out either in-house or externally, policy departments provide information in all areas of Parliament's activities. The study was undertaken upon request of the ENVI Committee of the European Parliament. The aim was to investigate the feasibility and opportunity of introducing a harmonised EU approach concerning the assessment of the added therapeutic value of medicines in the European Union. The study sets out the general background to ATV and its use by EU Member States, and explains the different stages in the national decision-making process where ATV considerations play a role. Next, it provides a (non-exhaustive) mapping of practices and the current state-of-play in the 28 EU Member States, as well as an in-depth analysis of the use of ATV in six selected countries: Austria, France, Italy, Poland, Slovakia and Sweden. Based on the information and data collected, the study also provides legal and policy recommendations for streamlining the various national systems in place for ATV assessment towards a common EU approach.
The concept of ATV
The concept of ATV has been proposed in several instances as a way to measure the therapeutic advantages of new medicinal products. While there is no universally agreed definition, the study understands the ATV of medicinal products as the incremental "therapeutic value" brought by a new drug or intervention compared with the best available treatment options already on the market. The therapeutic value can be defined in terms of positive patient-relevant endpoints and relevant levels of effectiveness, efficacy and safety.
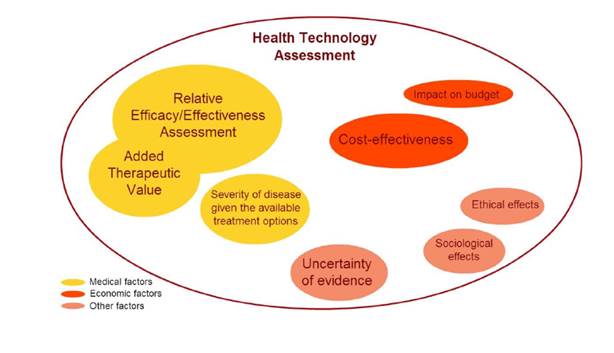
While the assessment of ATV could theoretically happen prior to the authorised entry of the medicine onto the market, in Europe it is conducted mainly by national authorities in charge of determining the pricing and reimbursement status of a new medicine. So called pharmaceutical benefit assessments (including an ATV assessment) play an increasingly important role in refunds by the national health services. In most European countries, a pharmaceutical has to successfully pass a post-approval benefit evaluation before it becomes available in the publicly funded healthcare system. Most European Member States have implemented health technology assessments (HTA)-based pharmaceutical evaluation schemes. HTA encompasses a systematic evaluation of the wider issues related to the introduction of a new medicine. This could include economic elements like cost-effectiveness and the impact on a healthcare budget as well as sociological and ethical effects such as the prevalence of the disease or its impact on a particular sub-group of patients. Criteria used to evaluate medicinal products are pictured as follows1:
ATV in the regulatory context
The current EU legal framework – mainly set out by Directive 2001/83/EC and Regulation 726/2004 – does not preclude single Member States from releasing marketing authorisations valid at national level, but it creates several routes for manufacturers to secure an EU-wide marketing authorisation. The most important of these is the "centralised authorisation procedure", which allows for a single marketing authorisation of a new medicinal product that is valid in all EU countries and Iceland, Liechtenstein and Norway. The centralised procedure is compulsory for certain medicinal products, for example, those manufactured using biotechnological processes. The procedure is managed by the European Medicines Agency (EMA), which conducts a benefit-risk assessment, balancing desirable and undesirable effects of the new medical product. The three criteria that are used during the assessment are: pharmaceutical quality, safety and efficacy. This also applies for the marketing authorisation procedures at national level. Market authorisation is granted in the case of a positive benefit-risk ratio, meaning that the expected benefits (e.g. efficacy, intended effect) outweigh the risks (e.g. safety concerns, unintended effects) when patients are exposed to the product.
However, the current EU legal framework does not require new medicinal products to be compared to the prevailing alternatives. Added therapeutic value is not systematically assessed in the marketing authorisation phase, nor are the manufacturers of the new medicine legally required to provide any other kind of comparisons to the prevailing treatment alternatives. Receiving a marketing authorisation is an essential, but only partial, condition for a new medicine to become available to patients in the EU.
Outcome of the study
 In the study, a mapping exercise was conducted of all EU Member States, and evidence of some form of ATV assessment being conducted was found in 26 countries. The measurement of ATV depends on the treatment the new medicine is compared with, and choosing one comparator over another may influence the end results. However, when looking at the definition of comparators used in EU countries, there is relatively little variation. Most countries identify "best standard care" as their main comparator of choice, even though some will allow and recommend additional comparisons with the "best possible care", or the "cheapest alternative". The evidence is mainly based on regulatory trials, and as a consequence the comparator used in those trials becomes a common assessment comparator for all countries, independent of the local use of that comparator.
In the study, a mapping exercise was conducted of all EU Member States, and evidence of some form of ATV assessment being conducted was found in 26 countries. The measurement of ATV depends on the treatment the new medicine is compared with, and choosing one comparator over another may influence the end results. However, when looking at the definition of comparators used in EU countries, there is relatively little variation. Most countries identify "best standard care" as their main comparator of choice, even though some will allow and recommend additional comparisons with the "best possible care", or the "cheapest alternative". The evidence is mainly based on regulatory trials, and as a consequence the comparator used in those trials becomes a common assessment comparator for all countries, independent of the local use of that comparator.
The study concluded that while there are differences in how ATV is assessed and defined across the EU Member States, the underlying principles are not fundamentally incompatible and share the same goals and concepts. As reasons for this conclusion the study names the results obtained in regard to the High Level Pharmaceutical Forum - a political platform set up by the European Commission in 2005 for a period of three years - and the Joint Action on Health Technology Assessment "EUnetHTA". The Forum underlined the importance of assessing the Relative Efficacy and Effectiveness of new medicinal products (REA) and endorsed REA's aim to compare healthcare interventions in daily practice and classifying them according to their added therapeutic value. It also recommended that Member States adopt the endorsed definition of REA, as the extent to which an intervention does more good than harm compared to one or more alternative, either under ideal circumstances (efficacy) or under the usual circumstance of health care practice (effectiveness). As another mechanism that has been put in place by the EU to facilitate cooperation on HTA between Member States, the Joint Action on Health Technology Assessment was mentioned. The purpose of this programme is to develop guidelines for effective joint working models for HTA collaboration as well as transparent governance tools, to prevent duplication of work between national agencies and to promote sharing and exchange of HTA information.2 The Cross Border Healthcare Directive further improved the conditions of information exchange between Member States by creating "a voluntary network connecting national authorities and bodies responsible for health technology assessment", which is now known as the Health Technology Assessment Network. To date, all Member States participate in this network, which consists of national authorities responsible for Health Technology Assessment.
The study clearly recommends a move towards a joint approach across Member States in the exercise of defining, measuring and estimating ATV. Agreeing on a shared definition of ATV is seen as "a desirable and logical next step". According to the policy department A, it should be possible for the Member States to agree on a shared definition and assessment methodology, as long as this is based on clinical criteria, rather than social and economic considerations. A shared definition and methodology to determine ATV would allow Member States to build on each other's expertise and make joint assessments easier. Therefore, it is recommended to strengthen further the cooperation between the various national competent authorities and EUnetHTA. According to the policy department A, the numerous interactions currently happening in the Joint Action programme might facilitate the development of a stepwise approach towards good practices for ATV assessment.
 Moreover, the study recommends the creation of an international European joint committee - composed of experts in the areas of medicine, clinical R&D, clinical pharmacology, biostatistics, epidemiology, niche expertise in specialised medical topics (e.g. auto-immune diseases, oncology etc.), and in rare diseases and advanced therapies - which performs the REA of medicinal products near to market authorisation, prior to local reimbursement submission and works independently from the financial, budgetary or economical assessments in the reimbursement track. Such a European joint committee in which each Member State delegates staff members should build on the exchange of expertise and good practices which currently takes place in the European Joint Action. This would bring together the EC, Member States and HTA agencies and the actual parallel assessment by competent authorities of similar clinical evidence included in the local reimbursement dossiers could be replaced by one joint assessment performed by the committee. Competent authorities at national level would be able to adopt the resulting joint REA reports, at their discretion. Concerning working procedures, the study recommends the inspiration of those from regulatory assessment committees such as the CHMP.
Moreover, the study recommends the creation of an international European joint committee - composed of experts in the areas of medicine, clinical R&D, clinical pharmacology, biostatistics, epidemiology, niche expertise in specialised medical topics (e.g. auto-immune diseases, oncology etc.), and in rare diseases and advanced therapies - which performs the REA of medicinal products near to market authorisation, prior to local reimbursement submission and works independently from the financial, budgetary or economical assessments in the reimbursement track. Such a European joint committee in which each Member State delegates staff members should build on the exchange of expertise and good practices which currently takes place in the European Joint Action. This would bring together the EC, Member States and HTA agencies and the actual parallel assessment by competent authorities of similar clinical evidence included in the local reimbursement dossiers could be replaced by one joint assessment performed by the committee. Competent authorities at national level would be able to adopt the resulting joint REA reports, at their discretion. Concerning working procedures, the study recommends the inspiration of those from regulatory assessment committees such as the CHMP.
The study, however, emphasised that while the Treaty on the Functioning of the European Union gives the EU legislative competences to set high standards of quality and safety for medicinal products, Member States are responsible for determining the resources assigned to health services and medical care. In general, in the field of public health, the EU plays only a role in supporting, coordinating and supplementing Member State actions. Hence, the study clearly makes a distinction in regard to the national competence on reimbursement decisions. The competence to accept or refuse the joint assessment report should remain with each Member State. Nevertheless, the study points out that the European Court of Justice has repeatedly clarified that, in exercising this competence, Member States must respect EU law in the public health field as well as other fields, notably competition.3 Thus, Member States may not undertake pricing/reimbursement policies or practices that unjustifiably distort competition. Besides, the Commission has already shown concern about the lack of transparency and the existence of discrimination in this area4. However, a proposal for a directive5 to improve the transparency of measures to regulate the price of medicinal products and their inclusion in public health insurance systems was withdrawn in March 2015. Nevertheless, the study pointed out the need for transparency in reimbursement decision-making which is "of utmost importance".
Assessment and outlook
The proposals of the policy department A in regard to future joint assessments tend to have the benefit of concentrating expertise that is not usually found within a single national agency and, once there is an agreement on their methodology, saving time. Moreover, the existence of a shared methodology would reduce the administrative burden for pharmaceutical manufacturers, who would need to invest fewer resources in tailoring their dossiers to each Member State where they wish to apply for reimbursement.
A shared definition of ATV is needed. This would clarify the expected benefits of new medicinal products, incentivising the production of innovative medicines. Whatever the level of harmonisation achieved is the REAs including the ATV estimation should be more transparent, in particular through detailed methodological guidelines that are made publicly available. This also gives the chance to allow appeals of pharmaceutical entrepreneurs on matters of content rather than just form.
 However, when it comes to more complex Health Technology Assessments, including social, ethical and economic considerations, the study concludes that harmonisation is more complex. Member States may have different opinions on the weight given to different factors, not to mention the fact that the data itself and the choices in healthcare spending will likely vary from one country to another. Nevertheless, the study proposes to share best practices and build staff capacities also in this area to achieve at least some kind of harmonisation level.
However, when it comes to more complex Health Technology Assessments, including social, ethical and economic considerations, the study concludes that harmonisation is more complex. Member States may have different opinions on the weight given to different factors, not to mention the fact that the data itself and the choices in healthcare spending will likely vary from one country to another. Nevertheless, the study proposes to share best practices and build staff capacities also in this area to achieve at least some kind of harmonisation level.
Importantly, the policy department A emphasised that whatever the level of harmonisation achieved, the pricing and reimbursement decision will remain the full competence of national authorities, following the EU HTA network's vision: "evidence is global, decision is local".
If you have any questions on this article or would like to propose a subject to be addressed by Synapse please contact us.
2 For more information see Health technology assessments and pharmaceutical pricing
3 See, for example, case C-372/04, Watts [2006] ECR I-4325; case C-169/07, Hartlauer [2009] ECR I-1721; case C-531/06, Commission v Italy [2009] ECR I-4103; case C-173/09, Elchinov [2010] ECR I-8889.
4 See the competition inquiry into the pharmaceutical sector


Related articles
![]() The Innovation Pathway - from R&D to you and me
The Innovation Pathway - from R&D to you and me
![]() Infringement of second medical use patents - important developments in Germany
Infringement of second medical use patents - important developments in Germany
![]() Managing IT projects in the healthcare sector - a European perspective
Managing IT projects in the healthcare sector - a European perspective
Previous articles
![]() Connected Health: regulatory issues
Connected Health: regulatory issues
![]() Abusive denigration in the pharmaceutical industry
Abusive denigration in the pharmaceutical industry
![]() Medical devices: liability of notified bodies?
Medical devices: liability of notified bodies?
![]() Connected health: Legal aspects of health apps in Germany
Connected health: Legal aspects of health apps in Germany
"The concept of ATV has been proposed in several instances as a way to measure the therapeutic advantages of new medicinal products."
"Agreeing on a shared definition of ATV is seen as 'a desirable and logical next step'."