The Medical Devices Regulation is coming!
July 2016
In 2012, the Commission published two regulation proposals to revise existing legislation on medical devices1 and in vitro diagnostics2. The revision affects all kinds of medical devices, from home-use items like pregnancy tests and contact lenses, to X-ray machines, pacemakers and breast implants. After nearly four years of negotiation between the EU institutions, industry and Member States, the end of the legislative procedure is almost in sight.
When does the new framework come into effect?
At first, it is important to note that, as an EU regulation, the Regulation on medical devices (MDR) will have the force of law throughout the EU when it comes into effect. While this will eliminate country-by-country interpretations of the requirements possible under the current directives, it might also speed up the implementation of the MDR’s requirements across the EU.
In regard to the legislative procedure, the European Council has finalised its position and in autumn 2015 entered into Trilogue – informal talks with the other EU institutions, i.e. the European Parliament and European Commission. This is a process that has been beset by delays, but the Dutch Presidency (incumbent from January to June 2016) has said that the proposed legislation could be published in the middle of the year. On 27 June 2016 the Council of the European Union published the latest status of the regulations. Presumably, on this basis an agreement will be reached and these will be the final texts. According to the just published documents, the MDR as well as the regulation on in vitro diagnostic medical devices will enter into force on the twentieth day after its publication in the Official Journal of the European Union. After that, the MDR will be in general applicable three years after its entry into force (Art. 97 para. 2), while a five-year period is proposed for the Regulation on in vitro diagnostic medical devices (Art. 90 para. 2). With regards to Brexit, presumably effective by then, the new regulations will not apply in the United Kingdom.

Significant changes
Therefore, manufacturers of medical devices will be confronted with major changes in the European Union’s regulatory framework which governs market access to the EU for medical devices. The proposed MDR differs in several important ways from the EU’s current directives for medical devices. Most probably, significant changes in the proposed regulation will include:
- More stringent clinical evidence: The MDR will require device manufacturers to conduct clinical performance studies and provide evidence of safety and performance proportionate with the risk associated with a given device. Device manufacturers will also be required to collect and retain post-market clinical data as part of the ongoing assessment of potential safety risks (Chapter VI MDR- clinical evaluation and clinical investigations).
- Responsible person: Device manufacturers will be required to have available within their organisation at least one person responsible for regulatory compliance who possesses the requisite expertise in the field of medical devices. The organisation must document the specific qualifications of this individual relative to the required tasks (Art. 13).
- European Databank on medical devices ("Eudamed"): Manufacturers of medical devices will have to fit their products with a unique device identification to allow the identification and facilitate the traceability of devices, other than custom-made and investigational devices (Art. 24).
- Scrutiny mechanism: According to Art. 44 a mechanism for scrutiny of conformity assessments of certain class III and class IIb devices were (again) included. Notified bodies must notify the competent authorities of certificates they have granted to class III devices and class IIb active devices intended to administer and/or remove a medicinal product. A competent authority and the Commission may, based on reasonable concerns, then apply further procedures.
- Intensified market surveillance: The position of notified bodies vis-à-vis manufacturers will be strengthened, including their right and duty to carry out unannounced on-site audits and to conduct physical or laboratory tests on medical devices to ensure continuous compliance by manufacturers after receipt of the original certification. Besides, the competent authorities can perform appropriate checks on the conformity characteristics and performance of devices and can, in this regard, carry out both announced and, if necessary, unannounced inspections of the premises of economic operators, as well as suppliers and/or subcontractors (Art. 67).
- Common specifications: The MDR plans to allow the EU Commission to publish common specifications which shall then be taken into account by manufacturers as well as notified bodies. These common specifications shall exist in parallel to the harmonized standards (Art. 7). Devices which are in conformity with the common specifications are presumed to be in conformity with the requirements of the regulation.
What does the new framework mean for currently marketed medical devices?
The MDR will apply three years after entry into force (twentieth day after its publication in the Official Journal of the European Union). However, Art. 94 MDR contains transitional provisions. Certificates issued by notified bodies in accordance with Directives 90/385/EEC and 93/42/EEC prior to the entry into force of the MDR will remain valid until the end of the period indicated on the certificate, except for certificates issued in accordance with Annex 4 of Directive 90/385/EEC or Annex IV of Directive 93/42/EEC which will become void at the latest two years after the date of application of the MDR. Certificates issued by notified bodies in accordance with Directives 90/385/EEC and 93/42/EEC after the entry into force of the MDR will remain valid until the end of the period indicated on the certificate, which shall not exceed five years from its delivery. They shall however become void at the latest four years after the date of application of the MDR. Devices which were lawfully placed on the market pursuant to Directives 90/385/EEC and 93/42/EEC prior to the date of application of the MDR may continue to be made available on the market or put into service until five years after that date.
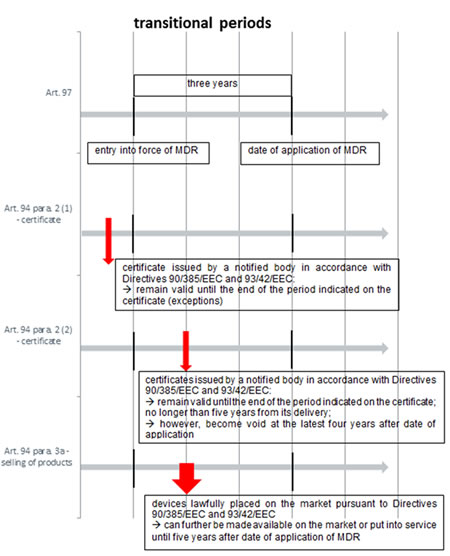
Besides, clinical investigations which have started to be conducted in accordance with Art. 10 of Directive 90/385/EEC or Art. 15 of Directive 93/42/EEC prior to the application of the MDR can be continued, however, the reporting of serious adverse events and device deficiencies shall then be carried out in accordance with the MDR.
How can manufacturers of medical devices prepare?
Additional changes to the MDR are still possible at this stage of the legislative procedure, and the actual terms of the proposed regulation are not final until the publication of the MDR in the Official Journal of the European Union. However, due to the new regulatory requirements and – in general – the necessity to obtain approval from a notified body, manufacturers of medical devices are well-advised to monitor the progress of the MDR as well as to consult their notified body. This is in particular important because currently approved medical devices are not exempt from the requirements of the MDR and will need to be re-evaluated and re-approved after the transitional periods. In this respect it needs to be considered that a large number of medical devices will require review and approval by a notified body again, therefore delays in the review and approval process by notified bodies should be expected. Advanced preparation and early action seems to be key to ensure a smooth transition to the new legal requirements.
If you have any questions on this article or would like to propose a subject to be addressed by Synapse please contact us.


Latest topical issues
![]() Damages in Trade Secrets Cases - "Derived Products"
Damages in Trade Secrets Cases - "Derived Products"
![]() Pemetrexed in Germany: Federal Court of Justice sets aside decision of Appeal Court
Pemetrexed in Germany: Federal Court of Justice sets aside decision of Appeal Court
Previous articles
![]() European Council approves Trade Secrets Directive
European Council approves Trade Secrets Directive
![]() Threats to sue for patent infringement - the risks
Threats to sue for patent infringement - the risks
![]() Attention returns to decisions on UPC opt-out
Attention returns to decisions on UPC opt-out
![]() Life Sciences Issues in Corporate Transactions - Part 1
Life Sciences Issues in Corporate Transactions - Part 1
![]() Life Sciences Issues in Corporate Transactions - Part 2
Life Sciences Issues in Corporate Transactions - Part 2
![]() A go-to guide to the Unified Patent Court and Unitary Patent
A go-to guide to the Unified Patent Court and Unitary Patent
"manufacturers of medical devices will be confronted with major changes in the European Union’s regulatory framework which governs market access to the EU for medical devices."
"currently approved medical devices are not exempt from the requirements of the MDR and will need to be re-evaluated and re-approved after the transitional periods"